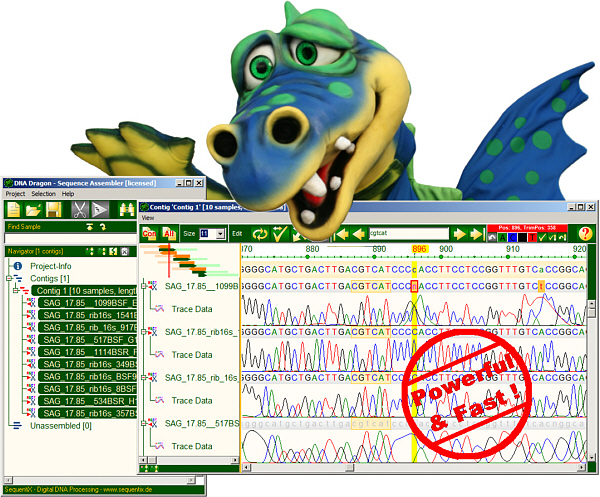
DNADragon
DNA Sequence Assembly Software
DNADragon is a desktop DNA sequence assembler software. It assembles partial sequences into contigs: automatic trimming, fast sequence assembly with base editing and proofreading. Supports next generation sequencing formats. It is especially useful for small to intermediate sequencing projects.
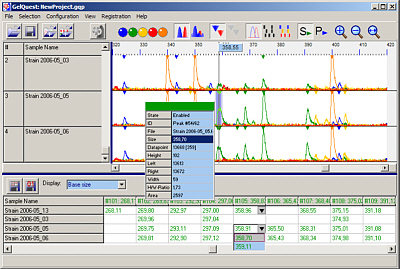
GelQuest/ClusterVis
Molecular Fingerprinting
Gelquest & ClusterVis are programs for the analysis of molecular fingerprint data: AFLP, t-RFLP, RFLP, RAPD, PFGE. Automatic peak detection and evaluation, binning, binary matrix construction, export of 01 matrix and other features. ClusterVis performs binary matrix analysis: distance matrix calculation, bootstrapping, cluster analysis, tree construction ...

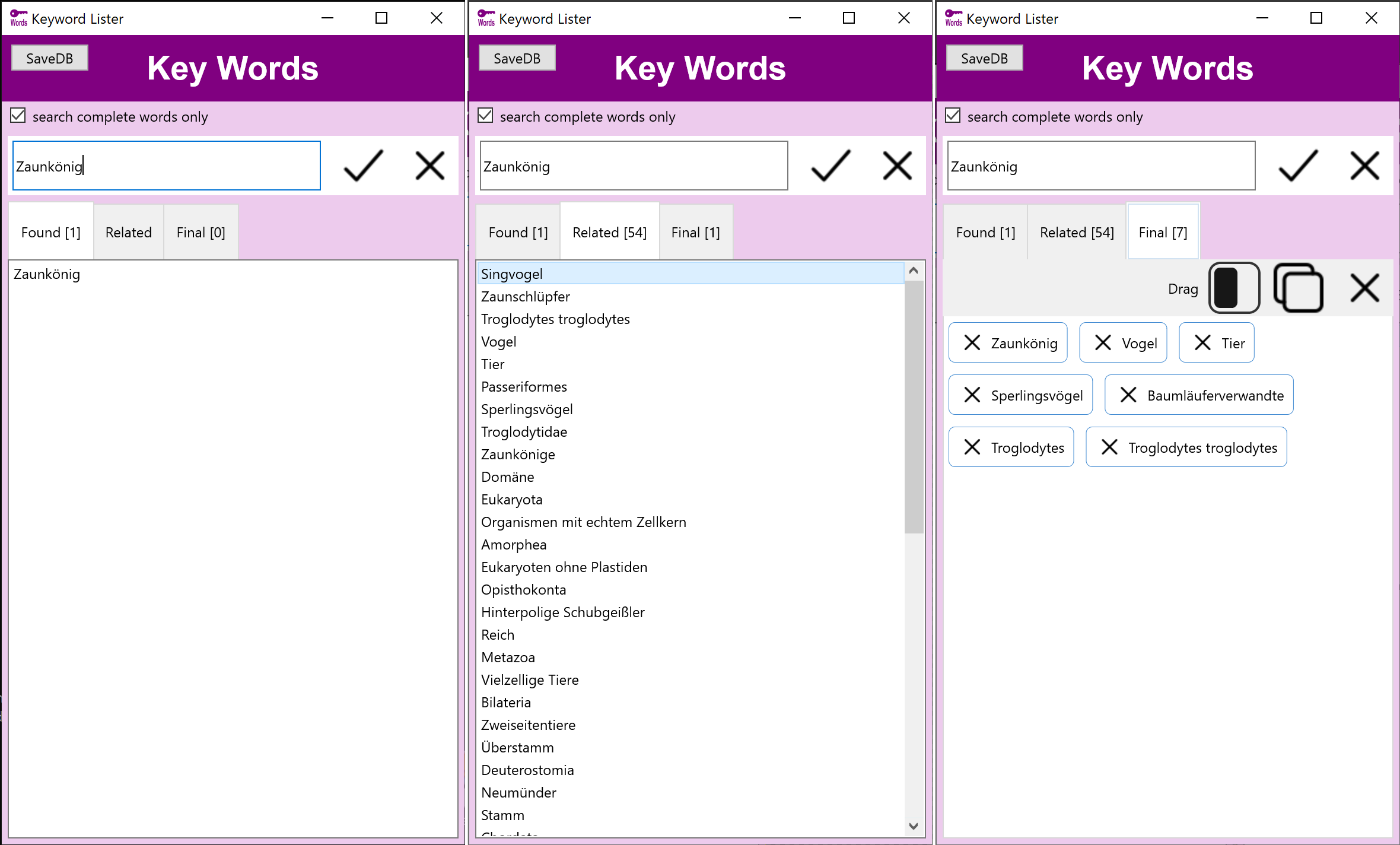
Stockmedia Schlagwort Tool - Deutsch
Schlagwörter, verwandte Wörter und Assoziationen
Stockmedia Schlagwort Tool - Deutsch hilft schnell die richtigen Schlagwörter und verwandte Begriffe zu finden und nach Priorität zu sortieren.
Die deutsche Version beinhaltet eine Datenbank mit mehr als 350000 Begriffen.
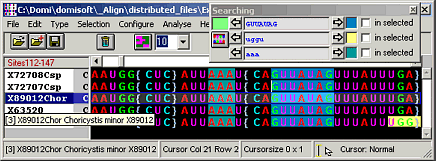
Align
Manual sequence alignment editor
Align is a manual DNA sequence alignment editor for DNA, RNA, and protein sequences.
Imports and exports various sequence file formats, e.g. Paup-Nexus, Phylip, Fasta, EMBL, GenBank, DCSE, and others.
Find/Highlight sequence motives in your molecular sequence alignment (including wildcards ? or *)
Wizard for automatic processing of sequence alignments for phylogenetic analyses
Define phylogenetic masks: Which sites are good for phylogenetic analysis?
Select/Deselect sequences and save selection profiles to define subsets.
Sort sequences by: accession numbers, taxonomy, arrangement in a phylogentic tree and others
Several cursor shapes: normal, exclusive (edits all sequences except cursor position), ex- and insertional, left-ended cursor
Change the cursor to edit several sequences simulataneously
Analyze sequences and alignments: similarities, p-distances, RASA, ...
Manipulate aligned sequences: Translate DNA/RNAs to protein using your own codon usage table
Cut aligned sequences: e.g. remove common positions, where a mask sequence contains special characters
Create sequences: e.g. consensus sequences, frequency of characters, sites that are only present in selected species (signatures), ...
Full access to all GenBank fields
Batch replacement of accession numbers by other fields in foreign files, e.g. replace labels in a tree file. This is not necessary if you have TreeMe.
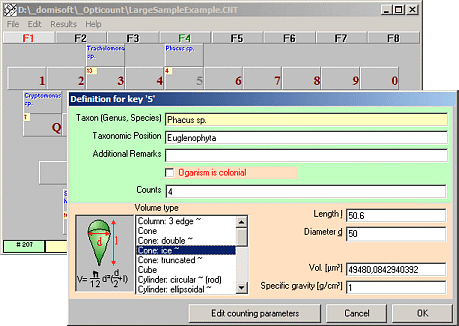
Opticount
Counts plankton and particles
Opticount is a user friendly software for the quantification of phytoplankton, zooplankton and bacterioplankton and even blood cells, particles and other things that cannot be counted automatically.
Organisms can be freely defined (genus and/or species names).
Support for counting unicellular and colonial organisms.
Individual cell volumes can be calculated from standard geometric objects or using fixed volumes. Volume biomass is automatically converted into fresh weight biomass.
Opticount automatically converts counts for commonly used counting procedures into cell concentrations. You may count organisms in counting chambers or concentrated on filter surfaces using five different methods:
1. lanes through the center,
2. randomly selected rectangular fields,
3. randomly selected circular fields,
4. percent area, or
5. using a blood counting chamber.
Just use a computer keyboard near to your microscope instead of a mechanic counting device.
Count single cells and/or colonies.
Species labels and actual counts are displayed..
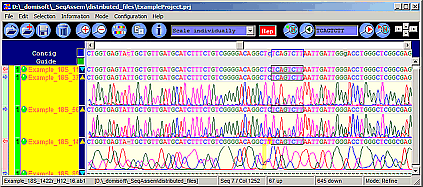
SeqAssem
Contig Sequence Assembly Software
SeqAssem - View, edit and proofread DNA sequences and on-the-fly assembly of partial sequences into contigs
Imports files of the following formats: SCF 2.0, 3.0, ABI, AB1, FASTA.
Saves all information in small project files.
Shows estimation of total sequence quality (bad, intermediate, good).
Sorts sequences by quality, contig number, file name, ... or just arrange sequences by drag & drop.
Edit bases and compare with their peaks in the electropherograms.
Original trace data files are not altered.
Small-sized project files: electropherogram data is excluded from project files.
Restore original sequences by a single mouse click!.
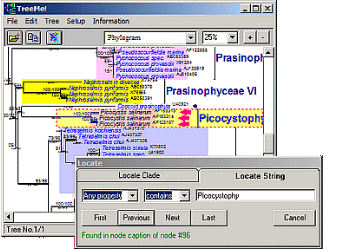
TreeMe
Visualization, Editing and Annotation of Phylogenetic Trees
TreeMe visualizes large phylogenetic trees (>1000 OTUs)
Combine trees: unites bootstrap labels of several trees onto a single tree
Full access to each node: modify node caption, branch caption, branch width, branch length, etc.
Annotate internal nodes (cluster nodes, clusters)
Collapse, Expand and Flip clusters
Re-root trees by using an outgroup
Define and visualise tags, e.g. arrows, boxes, text labels, etc.
Automatic replacing of taxon abbreviations (e.g. GenBank accession numbers) by complete names (e.g. by species names) using tab-delimited replacement files (edited with Microsoft EXCEL).
Fast location of nodes by searching for node names or other properties
Edit trees: move nodes, delete internal nodes, ...
Add links to taxa (e.g. to GenBank) and jump to the sequence data with a single mouse click!
Clipboard functions: Copy tree and insert into other software (Word, Powerpoint, ...)
Printing of trees
Imports: NEWICK (Phylip, MrBayes), NEXUS (Paup), ...
Outputs: NEWICK (Phylip, MrBayes), NEXUS (Paup), Vector Graphics (EMF, Windows Enhanced Metafile).
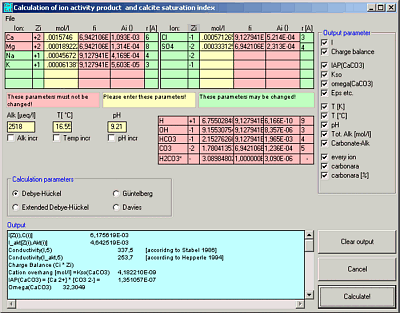
WinIAP
Ion Activities and Calcite Saturation Index
WinIAP calculates the ion activity products and the calcite saturation index for a given set of ions in a solution.
Enter, save and load ion concentrations.
Calculates ion activities and carbon species using 4 different equations (Debeye-Hückel, Extended Debeye-Hückel, Gürtelberg, Davies)
Calculates the calcite saturation index (S.I.).